Developmental mechanisms triggered by ncRNAs
Team REGARN / Martin Crespi
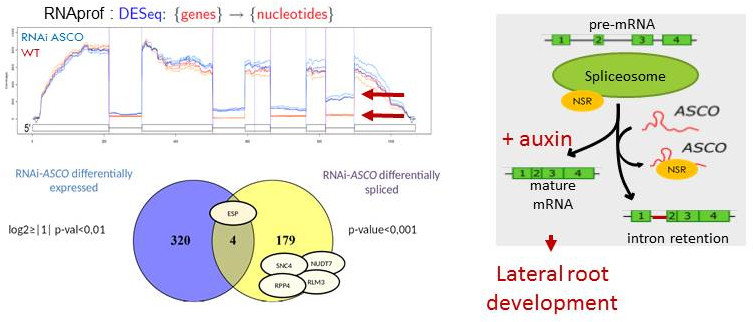
In this second theme, we aimed to better understand the mechanisms through which ncRNAs can affect development and the molecular basis of the interaction of ncRNAs with specific ribonucleoproteins in order to modify target gene expression. In the frame of the EPIRNA labex SPS project (coordinator M. Crespi), we have identified the ASCO lncRNA (for Alternative Splicing Competitor) that was able to interact with nuclear alternative splicing regulators named AtNSRa and AtNSRb (for Nuclear Speckle RNA-binding protein). Interestingly, by modulating the expression of this lncRNA, we modified the NSR-dependent alternative splicing patterns and the formation of lateral roots. We therefore proposed that modulation of alternative splicing could be a novel regulatory mechanism mediated by ncRNAs. These lncRNAs would "mimic" introns and may serve to modulate alternative splicing during development (Bardou et al., 2014; Van Du et al., 2015). This is an emerging mechanism in eukaryotes and there are growing evidences since then that lncRNAs can interfere with alternative splicing regulation (Romero-Barrios et al., 2018).
In addition to these “splicing-related” lncRNAs, we identified another group of lncRNAs exclusively co-localizing with 24nt siRNAs, known to be linked to epigenetic changes and gene silencing. Interestingly, extinction of one of these lncRNAs led to repression of its neighbouring gene PID (for PINOID kinase), a major regulator of auxin responses in roots. Then, we showed that this lncRNA, named APOLO (for Auxin PrOmoter-dependent LOop lncRNA), is able to regulate a "chromatin loop" spanning its position up to the target gene in order to control the epigenetic states of the PID target promoter. Analysing histone changes (in collaboration with Moussa Benhamed’s group, IPS2), as well as the status of DNA methylation and the formation of the chromatin loop in response to auxins, we found that APOLO may organize the chromatin structure of the PID promoter (Ariel et al., 2014). This work supports the notion that nuclear lncRNAs are key actors in the regulation of the 3D structure and epigenetic states of chromatin, opening up interesting perspectives in both plants and animals. We believe that this class of non-coding RNAs may be a new element involved in genome topology and chromatin structure (Ariel et al., 2015).
We also sought for changes in epigenetic reprogramming mediated by ncRNAs in Medicago truncatula. The understanding of the coordinated differentiation of plant and bacterial cells during the symbiotic interaction with Sinorhizobium meliloti is a developmental transition accompanied by massive reprogramming of gene expression that may involve epigenetic regulations. As mentioned certain epigenetic changes are associated with the production of 24nt siRNAs and APOLO-like lncRNAs (Ariel et al., 2014). In our previous work (Formey et al., 2014), we were puzzled that many siRNAs were specifically expressed during nodulation. In collaboration with Pascal Gamas’s lab and J. Gouzy (LIPM Toulouse) in the frame of the EPISYM ANR project (coordinator M. Crespi), we have identified that, in contrast to MtDCL3, the main enzyme involved in 24nt siRNA biogenesis, another RNase III (called RTL1 for RNAse-Three-Like 1) is drastically up-regulated in the differentiation zone of the nodules. These enzymes are likely capable to antagonistically modify in vivo the repertoire of 24nt siRNAs in M. truncatula nodules. Functional analyses both for Medicago DCL3 and RTL1 suggest an association with interesting symbiotic phenotypes. In particular, repression of RTL1 by RNAi strongly impaired nodule development, their ability to fix nitrogen and bacteroid viability.
Due to their role in fine tuning the expression of other genes, we expect that ncRNAs would modify quantitatively the observed phenotype either in intensity but also in timing. To help us, we are currently building a phenotyping robot in collaboration with the FabLab Digiteo. It will allow us to monitor more plants at a higher time resolution and therefore to capture fine modifications of root phenotype expected from ncRNAs expression modification.