Exploring the diversity of ncRNAs: linking ncRNAs to root plasticity
Team REGARN / Martin Crespi
In this first theme, we used a series of genomic approaches to identify and annotate both long and small ncRNAs mainly in M. truncatula and A. thaliana and more recently in other species in collaboration with different consortia, in order to detect potential ncRNAs associated with changes in root growth response. We have performed a global analysis of mi/siRNAs in M. truncatula (conserved and novel ones, both legume- or Medicago-specific) in the context of the MIRMED project (coordinator M. Crespi) where 23 different growth conditions of roots were used. This allowed us to analyse the small RNAs networks in this model legume, including new targets for conserved miRNAs as well as new miRNAs, notably in the symbiosis context (Formey et al., 2014, Lelandais-Brière et al., 2016). There is extensive knowledge of the genomes of M. truncatula accessions, being developed by Nevin Young (and international collaborators, including J. Gouzy, F. Débelle and L. Gentzbittel in France). Interestingly, genome-wide association studies (GWAS) performed in N. Young's laboratory to identify novel genes involved in the adaptation of the symbiosis to different environments showed that the majority of the identified loci correspond to intergenic regions. Hence, the analysis of miRNA/siRNA/lncRNA polymorphisms could be very relevant in this context in Medicago spp. More recently, we were involved in a collaboration project to use PACBIO technology for detailed sequencing of the M. truncatula genome which allowed the detection of “symbiotic islands” where a large number of lncRNAs were observed as “embedded” in coregulatory networks with symbiosis genes (Pécrix et al., Nature Plants in press). We have also analysed miRNA/siRNA diversity in a large collaborative project in sunflower linked to the full sequencing of this giant genome (Badouin et al., 2017).
In addition to the identification, we started the functional analysis of certain miRNAs in M. truncatula. We could show that miR160 and miR396 regulate root growth, auxin response and mycorrhization and highlighted the impact of RDR6 and the related phasiRNAs in many aspects of plant development (Bustos-Sanmamed et al., 2013, Bazin et al., 2013, Bustos-Sanmamed et al., 2014). More recently, we analysed the action of new miRNAs identified as exclusively expressed in the root apex (Formey et al., 2014). Inhibition of the action of one of these miRNAs strongly prevented root growth repression by cadmium. This miRNA cleaves a lncRNA to produce secondary phasiRNAs which may target specific genes limiting root growth in response to cadmium in sensitive phenotypes, as we could show by testing the rdr6 mutant impaired in phasiRNA but not in tasiRNA pathway (Bustos-Sanmamed et al., 2014). The fact that this new miRNA was also differentially induced between ecotypes showing contrasting root growth responses to cadmium further strengthens a potential link with ecotype adaptation. We propose that this regulatory network involving a new miRNA and targeted lncRNAs producing phasiRNAs, modulates the response of Medicago roots to cadmium, a very interesting trait (Proust et al., in preparation).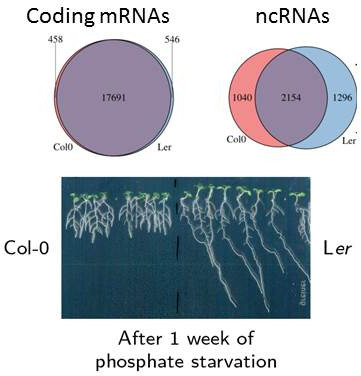
In Arabidopsis thaliana, we focused our efforts on the root adaptation to phosphate starvation. In this species, low concentration of inorganic phosphate (low-Pi) reduces primary root growth to the benefit of lateral roots. Interestingly, a QTL approach revealed the LPR genes (LOW PHOSPHATE RESPONSIVE) whose expression in the root cap is required for low Pi response. This and other data suggest that Pi sensing at the root apex is critical to modify root architecture during phosphate starvation. We started, in 2013, a collaboration with L. Nussaume's laboratory (CEA, Cadarache, France) to address the evolution of ncRNA networks in the root apex among ecotypes (Col-0 and Ler) showing differential root growth responses to low-Pi (ANR RNADAPT project). Using RNA sequencing, we compared complete transcriptional response (mRNAs, lncRNAs and small RNAs) of root tips from these two ecotypes. We identified thousands of new lncRNAs conserved at the DNA level. However, in contrast to coding genes, many lncRNAs were specifically transcribed in one ecotype (see Figure showing the number of ecotype-specific expression of coding mRNAs and ncRNAs). These ecotype-specific lncRNAs were further characterized by analysing their variability among sequenced Arabidopsis ecotypes and their ability to generate siRNA. A majority of lncRNAs were differentially regulated according to genotype rather than phosphate starvation, suggesting a role in ecotype adaptation. We performed several screens (such as co-regulatory patterns with neighbouring genes or antisense interactions with known critical regulators) to link these ecotype-specific lncRNAs to the differential root growth response between ecotypes. Functional analysis revealed that miss-expression of two ecotype-specific lncRNAs affects primary root growth in Col-0 (Blein et al., in preparation). Concerning miRNAs, we continued the analysis of miRNA action on specific transcription factors involved in root architecture such as miR169 and NFYA2. We demonstrated that specific miR169 isoforms, carrying point mutations compared to the canonical miR169 sequence, were involved in lateral root (LR) formation and primary root growth (Sorin et al., 2014).
Another emerging question in the ncRNA field is whether these new lncRNAs are really non-coding or may code for small peptides? To address this question, we established collaboration with Julia Bailey Serres (UCSF Riverside, USA) in the frame of an outgoing Marie Curie Fellowship. We determined ribosome footprinting of polyadenylated RNAs in roots of Arabidopsis shifted from high Pi to low Pi. We found, out of 2382 potential lncRNAs, that nearly one half were associated with ribosomes. Small ORF (sORF) prediction, calculation of ribosome termination and peptide analysis revealed that many sORFs are hidden in lncRNAs and may produce small peptides. Indeed, we identified 34 evolutionarily conserved and conditionally regulated peptides encoded in lncRNAs. In addition, for natural antisense transcripts (NATs), sORFs with ribosomes footprints predominated near the 5'-termini of transcripts. These included Pi-upregulated NATs whose activation correlated with sense mRNA Pi-enhanced translation. Furthermore, our data provide evidence for translation of well-known lncRNAs such as trans-acting and phased siRNAs (tasiRNA and phasiRNAs) genes and a miRNA target mimic although not of miRNA precursors. Through mutational analyses, we showed that translation of a non-conserved sORF in the TAS3a gene enhances the biogenesis of the related tasiRNAs. Altogether, we propose that sORF translation may determine the stability and function of certain lncRNAs (Bazin et al., 2017). The developed datasets in this context allowed us to further classify the previously mentioned lncRNAs (when detected in Col-0) based on their ribosome association.
In parallel to this work, we continued to study the interaction between the mechanisms of degradation by mi/siRNAs and those involved in the quality control of mRNAs (RQC or RNA quality control; Moreno et al., 2013). We showed that P-bodies and siRNA-bodies act antagonistically on similar “aberrant” RNAs. These adjacent "bodies" revealed new aspects of the subcellular dynamics of RNA degradation (Martinez-Alba et al., 2015). We are extending this work using several RQC and splicing mutants (in Col-0 background) to further assess the dependency of lncRNA expression in ecotypes (e.g. the Ler ecotype carries a mutation in the SOV gene involved in RNA quality control, RQC). This may allow us to further explore links between the evolution of new regulatory pathways in ecotypes through mutations in RQC or splicing pathways.