- Présentation
- Recherche
- FunRNA : Conservation fonctionnelle des longs ARN non codants des plantes
- FLOCAD : Développement floral et déterminisme du sexe
- Qlab : Equipe Génomique et épigenomique quantitative des plantes
- ChromD : Dynamique des chromosomes
- SILAB : Voies de signalisation régulant l'architecture du système racinaire des légumineuses en réponse à des bactéries bénéfiques
- CCARS : Changement climatique et signalisation redox
- STRESS : Voies de signalisation du stress
- OGE : Expression des génomes des organites
- Modélisation mathématique et informatique pour les études plant-omics COMPAS / Marie-Hélène Mucchielli-Giorgi & Guillem Rigaill
- GUILLOTIN Lab
- Enseignement
- Plateformes
- Bases de données
Axes de recherche
Pour atteindre ces objectifs, les principaux axes de recherche de l’équipe COMPAS visent à :
• Analyser des données omiques sans annotations de référence complètes (coordination : Guillem Rigaill)
La régulation de la transcription est un processus complexe, essentiel au fonctionnement et au développement de tous les organismes vivants. Elle contrôle l'expression des gènes et joue un rôle fondamental dans de nombreux processus biologique tels que la différenciation cellulaire, le développement, l'homéostasie, la réponse aux stimuli et l’apparition de certaines maladies.
Les analyses RNA-seq classiques, utilisées pour étudier les événements de régulation, passent cependant à côté d’une part importante de ces phénomènes. En effet, la plupart des approches reposent uniquement sur des transcrits bien caractérisés obtenus à partir de bases de données de référence. Les transcrits non-référencés sont ignorés et cela entrave la compréhension moléculaire des phénomènes biologiques.
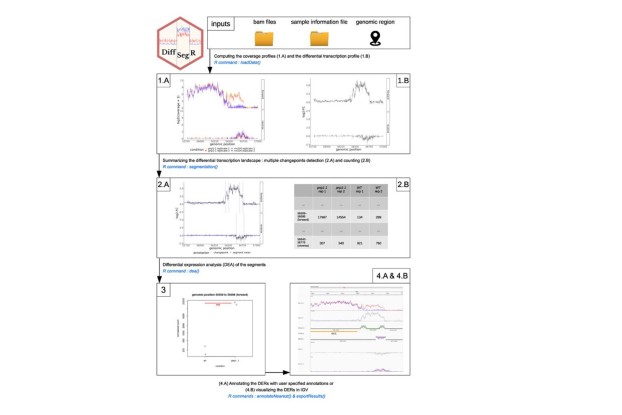
Nous développons des approches capables de détecter des régulations transcriptomiques sans caractérisation préalable des transcrits. Ces régulations correspondent à des régions, petites ou grandes, différentiellement exprimées, le long du génome. Formellement, détecter ces régions est un problème de détection de ruptures ou de segmentation.
L’approche DiffSegR [Liehrmann et al., 2023] que nous avons développé identifie la majorité des événements de régulation dans de petits génomes (chloroplaste) tout en contrôlant le nombre de faux positifs. Nous souhaitons à présent étendre cette stratégie à des génomes plus grands. Cela passe par des développements en statistiques et apprentissage pour permettre l'inférence de modèles de détection de ruptures complexes sur de très grands génomes. Nous effectuons ces développements avec des collègues statisticiens et mathématiciens en France et à l’étranger (voir: http://www.math-evry.cnrs.fr/members/grigaill/welcome).
Lien bibliographique :
Liehrmann A et al. (2023) https://doi.org/10.1093/nargab/lqad098
• Inférer et analyser des réseaux d’interactions protéine–protéine (coordination : Marie-Hélène Mucchielli-Giorgi).
Les réseaux d’interactions protéine-protéine (PPI) constituent une source majeure d’informations biologiques. Nous développons des approches pour inférer et analyser ces réseaux afin d’en extraire de nouvelles connaissances.
Nous avons notamment conçu APPINetwork [Gosset et al., 2022] (https://forge.inrae.fr/GNet/appinetwork), un outil permettant de construire et d’analyser des réseaux de PPI à partir de bases de données publiques, de données expérimentales non publiées et d’interactions prédites. Les approches d’analyse fondées sur les propriétés des graphes permettent d’identifier les protéines clés (hubs), de caractériser des modules fonctionnels [Glatigny et al., 2017] et de modéliser la biogenèse de grands complexes protéiques [Glatigny et al., 2011].
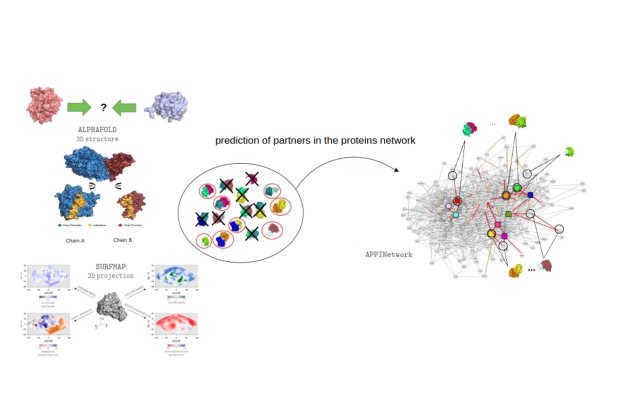
Cependant, les réseaux de PPI restent partiels et incomplets, limitant la compréhension globale des processus biologiques. Nous développons donc en parallèle des méthodes de prédiction d’interactions afin de compléter et filtrer ces réseaux. Si les approches actuelles (docking moléculaire, apprentissage profond) prédisent efficacement la structure 3D des complexes, elles ne permettent pas toujours de déterminer si deux protéines interagissent réellement.
Nous avons ainsi récemment développé une méthode innovante de prédiction des interactions chez Arabidopsis thaliana [Gosset, 2024], basée sur les critères de qualité fournis par AlphaFold2-Multimer. La stratégie consiste à prédire la structure du complexe, puis à combiner, via une approche de machine learning, différents scores de qualité et des descripteurs d’interface afin d’estimer une probabilité d’interaction.Les propriétés de surface sont calculées à l’aide de SURFMAP [Schweke et al., 2022],(https://github.com/i2bc/SURFMAP), un outil que nous avons développé pour analyser les surfaces protéiques.
Les performances obtenues sont très prometteuses : environ 70 % des interactions sont correctement prédites pour seulement 5 % de faux positifs. Cette méthode est désormais intégrée dans plusieurs projets collaboratifs au sein de l’IPS2 afin d’identifier les partenaires potentiels de protéines d’intérêt. Une application web permettant d’estimer automatiquement la probabilité d’interaction à partir des modèles structuraux prédits est actuellement en cours de développement.
Nous souhaitons à présent étendre cette stratégie à la prédiction des interactions entre facteurs de transcription, en utilisant AlphaFold3 pour modéliser les complexes formés par deux facteurs de transcription en présence d’ADN.
Liens bibliographiques :
Gosset et al. (2022) https://doi.org/10.7717/peerj.14204
Gosset (2024) https://theses.hal.science/tel-04540573
Glatigny et al. (2017) https://doi.org/10.1186/s12918-017-0442-0
Glatigny et al. (2011) https://doi.org/10.1186/1752-0509-5-173
Schweke et al. (2022) https://doi.org/10.1021/acs.jcim.1c01269
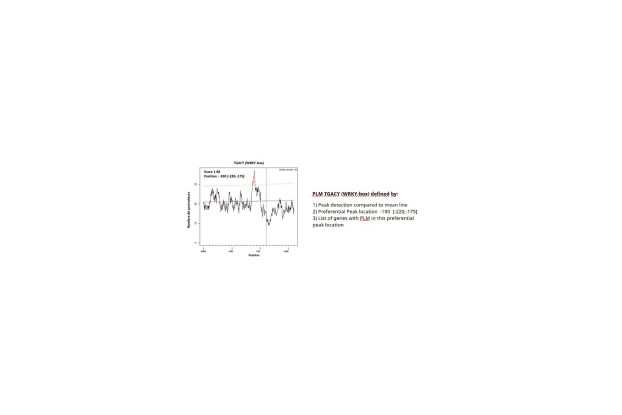
Identifier et caractériser des éléments cis-régulateurs (CRE) (coordination : Véronique Brunaud)
Les facteurs de transcription (TF) jouent un rôle clé dans la régulation de la transcription pour activer ou réprimer l’expression de gènes. Bien que nous connaissions les mécanismes généraux de leur action, notamment leur fixation à l’ADN au niveau des régions promotrices, les modalités précises de cette régulation demeurent largement mal caractérisées, en particulier concernant l’identification des TF impliqués pour un gène donné et les combinaisons de facteurs nécessaires à son contrôle.
Dans l’équipe COMPAS, nous avons développé la méthode PLMdetect [Bernard et al. 2010], qui fait l’hypothèse que (1) les motifs ADN cibles de TF sont en majorité dans des régions proximales des gènes et (2) que ces motifs sont à une distance spécifique du début de gène. Ces motifs prédits sont appelés PLM pour « Preferentially Located Motifs ». Lors d’un travail de thèse, il a été démontré que 90% des PLMs étudiés correspondaient effectivement à des sites de fixation de facteurs de transcription. . Cette validation a été réalisée en comparant la localisation des PLM avec des données expérimentales de liaison des TF obtenues par ChIP-seq et DAP-seq
Cette méthode a été appliquée (1) chez Arabidopsis sur des listes de gènes d’intérêt (par exemple des gènes coexprimés) [Bueso et al. 2014, Bueso et al. 2016, Del Prete et al. 2019], (2) de façon plus exploratoire chez le maïs [Rozière et al. 2022], et (3) à la recherche de motifs régulateurs de la traduction dans les organites cellulaires [Tran et al. 2023].
Sachant que l’expression des gènes résulte généralement de l’action combinée de plusieurs facteurs de transcription (TF), nous travaillons actuellement sur un projet portant sur la co-présence de PLM. Nous avons également développé une interface web, Plante-PLMview, pour faciliter l’accès à PLMdetect pour 20 espèces de plantes modèles et cultivées. (https://plmview.ips2.universite-paris-saclay.fr)
Liens bibliographiques :
Bernard et al. (2010) https://doi.org/10.1139/g10-042
Bueso et al. (2014) https://doi.org/10.1104/pp.113.232223
Bueso et al. (2016) https://doi.org/10.1111/tpj.13220
Del Prete et al. (2019) https://doi.org/10.1186/s12870-019-1738-6
Rozière et al. (2022) https://doi.org/10.3389/fpls.2022.976371
Tran et al. (2023) https://doi.org/10.1126/science.adg0995